Thermal
conductivity detector ที่เรียกกันย่อ
ๆ ว่า TCD
(หรือในชื่อเก่าว่า
Katharometer)
เป็นอุปกรณ์วัดตัวหนึ่งที่มีการใช้งานกันอย่างแพร่หลาย
ไม่ว่าจะเป็นในเรื่อง การวัด
องค์ประกอบ ความเร็ว และอุณหภูมิ
ของแก๊สที่ไหลผ่านตัว TCD
ที่เปลี่ยนแปลงไป
การทำงานของ TCD
อาศัยการเปรียบเทียบความแตกต่างระหว่างความสามารถในการระบายความร้อนออกจากขดลวดความร้อน
ในงานที่ใช้วัดองค์ประกอบของแก๊สที่ไหลผ่านว่าเปลี่ยนแปลงหรือไม่นั้นจะใช้ขดลวดสองขด
โดยขดหนึ่งนั้นเป็นตัวอ้างอิง
(reference)
และอีกขดหนึ่งนั้นเป็นขดลวดที่ให้แก๊สที่ต้องการวิเคราะห์ไหลผ่าน
ปัจจัยที่ส่งผลต่อความสามารถในการระบายความร้อนออกจากขดลวดความร้อนได้แก่
๑.
อัตราการไหล
โดยแก๊สที่ไหลเร็วจะระบายความร้อนได้ดีกว่าแก๊สที่ไหลช้ากว่า
๒.
อุณหภูมิของแก๊ส
โดยแก๊สที่เย็นกว่าจะระบายความร้อนได้ดีกว่าแก๊สที่ร้อนกว่า
และ
๓.
องค์ประกอบของแก๊ส
โดยแก๊สที่มีน้ำหนักโมเลกุลที่ต่ำกว่าจะระบายความร้อนได้ดีกว่าแก๊สที่มีน้ำหนักโมเลกุลที่สูงกว่า
ในการวิเคราะห์องค์ประกอบนั้น
สิ่งที่เราต้องควบคุมให้คงที่คือ
อัตราการไหล และอุณหภูมิ
ของแก๊สที่ไหลผ่านขดลวดแต่ละขดของตัว
TCD
ทั้งนี้เพื่อให้สัญญาณที่ตัว
TCD
ส่งออกมานั้นเป็นผลที่เกิดจากการเปลี่ยนแปลงความเข้มข้นเพียงอย่างเดียว
ซึ่งเป็นเรื่องที่ทำได้ไม่ยากในกรณีของการวิเคราะห์ที่
"อุณหภูมิคงที่"
แต่มักจะมีปัญหาเมื่อทำการวิเคราะห์แบบที่มี
"การเปลี่ยนแปลงอุณหภูมิ"

รูปที่
๑ แผนผังอย่างง่ายของอุปกรณ์ที่ใช้
TCD
ในการวัดองค์ประกอบของแก๊สที่เปลี่ยนแปลงไป
รูปบนคือเครื่องแก๊สโครมาโทกราฟ
(gas
chromatograph หรือ
GC)
รูปล่างเป็นของอุปกรณ์พวก
Temperature
programmed techniques
ตัวอย่างเช่นในกรณีของเครื่องแก๊สโครมาโทกราฟ
(ดูรูปที่
๑ ประกอบ)
แก๊สที่ไหลเข้าขดลวดอ้างอิงและขดลวดที่เป็นตัววัดองค์ประกอบนั้นเป็นแก๊สคนละเส้นทางกัน
ในตอนปรับตั้งเครื่องนั้นเราสามารถที่จะตั้งให้อัตราการไหลของแก๊สทั้งสองสายนั้นเท่ากันได้
และถ้าคอลัมน์ที่แก๊สไหลผ่านนั้นมีความยาวเพียงพอ
ก็จะทำให้อุณหภูมิของแก๊สทั้งสองสายที่ไหลเข้า
TCD
นั้นเท่ากันได้
ซึ่งจะเท่ากับอุณหภูมิของตัว
oven
ที่ติดตั้งคอลัมน์ทั้งสอง
แต่ถ้าเราทำการวิเคราะห์โดยมีการเพิ่มอุณหภูมิคอลัมน์ให้สูงขึ้นระหว่างการวิเคราะห์
สิ่งที่มักจะเห็นกันก็ TCD
จะส่งสัญญาณออกมา
ซึ่งทำให้เส้น base
line มีการเปลี่ยนแปลงไป
(และมักจะทำซ้ำไม่ค่อยได้)
การที่
TCD
ส่งสัญญาณออกมานี้ไม่ได้เกิดจากแก๊สที่ไหลผ่านขดลวดทั้งสองมีองค์ประกอบที่แตกต่างกัน
แต่เกิดจากการที่แก๊สที่ไหลผ่านขดลวดทั้งสองนั้นมี
"อุณหภูมิ"
และ/หรือ
"อัตราการไหล"
ที่แตกต่างกัน
สัญญาณที่
TCD
ส่งออกมานี้เป็นผลมาจากการที่คอลัมน์ที่ใช้ในการวิเคราะห์และใช้กับสายอ้างอิงนั้นไม่เหมือนกัน
จึงทำให้แก๊สที่ไหลผ่านคอลัมน์ทั้งสองไม่ได้มีอุณหภูมิสูงขึ้นในอัตราเดียวกัน
แก๊สที่ไหลผ่าน TCD
จึงมีอุณหภูมิที่แตกต่างกัน
และด้วยการที่แก๊สนั้นเมื่อมีอุณหภูมิสูงขึ้นจะมีความหนืดขึ้น
จะทำให้ความเร็วของแก๊สที่ไหลผ่านคอลัมน์ทั้งสองนั้นลดต่ำลง
(ในกรณีที่ใช้การปรับความดันด้านขาเข้าเพียงอย่างเดียวในการปรับอัตราการไหล)
ตรงนี้ถ้าใครมีเครื่อง
GC
ที่ติดตั้งตัวตรวจวัดชนิด
TCD
ก็สามารถลองเล่นดูได้
โดยทดลองเพิ่มอุณหภูมิคอลัมน์ให้สูงขึ้นด้วยอัตราเร็วตามที่กำหนดโดยไม่มีการฉีดสารตัวอย่าง
แล้วคอยดูว่าสัญญาณที่ออกมานั้นมีการเปลี่ยนแปลงอย่างไร
เทคนิคการวิเคราะห์โดยมีการเพิ่มอุณหภูมิตัวอย่างให้เพิ่มขึ้นด้วยอัตราที่กำหนดในระหว่างการวิเคราะห์นั้น
(ที่เรียกว่า
Temperature
programmed technique) เพื่อดูการเปลี่ยนแปลงทีเกิดขึ้น
เป็นเทคนิคหนึ่งที่มีการใช้กันอย่างแพร่หลายในการศึกษาตัวเร่งปฏิกิริยา
แผนผังอย่างง่ายของอุปกรณ์ตระกูลนี้แสดงไว้ในรูปที่
๑ โดยตัวตรวจวัดที่มีการใช้กันอย่างแพร่หลายก็คือตัว
TCD
ในการออกแบบนั้นจะให้แก๊สไหลเข้า
TCD
ฝั่งขดลวดอ้างอิงก่อน
จากนั้นจึงให้แก๊สตัวนี้ไหลผ่านคอลัมน์บรรจุตัวอย่างที่ติดตั้งอยู่ใน
oven
ที่ควบคุมอุณหภูมิตัวอย่าง
แก๊สที่ไหลผ่านตัวอย่างออกมาอาจผ่านเข้า
cold
trap (จะมีการใช้หรือไม่ขึ้นอยู่กับรูปแบบการวิเคราะห์)
ก่อนที่จะไหลเข้าสู่ขดลวดที่สอง
สิ่งที่คาดหวังจะเห็นกันก็คือ
สัญญาณที่ TCD
ส่งออก
มานั้นควรเป็นผลจากการที่แก๊สที่ไหลเข้าขดลวดอ้างอิงกับที่ไหลออกมาจากตัวอย่างนั้นมี
"องค์ประกอบที่แตกต่างกัน"
เท่านั้น
แต่เอาเข้าจริงมันมักไม่เป็นเช่นนั้น
เพราะแม้แต่เราเอาวัสดุที่เฉื่อยบรรจุไว้ในคอลัมน์
แล้วทดลองเพิ่มอุณหภูมิคอลัมน์ให้สูงขึ้น
เราก็ยังเห็น TCD
ส่งสัญญาณออกมาอยู่ดี
ทั้ง ๆ ที่องค์ประกอบของแก๊สที่ไหลผ่านขดลวดทั้งสองเหมือนกัน
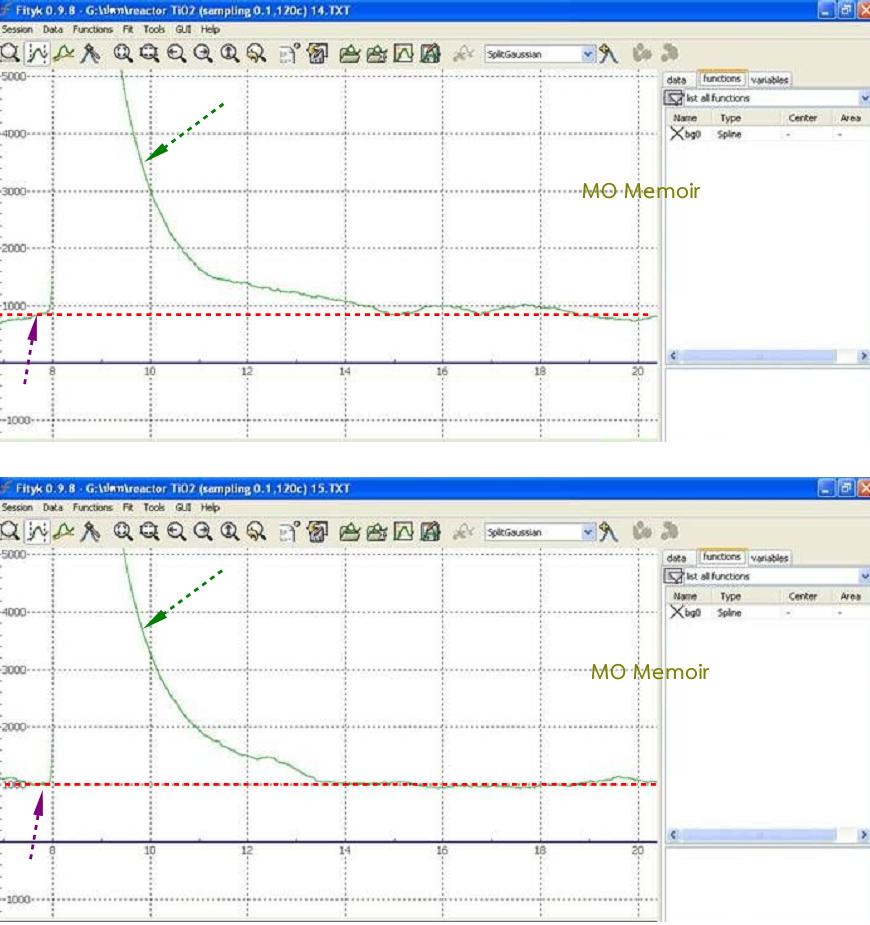
ดังเช่นตัวอย่างที่นำมาแสดงในรูปที่
๒ ที่ให้เฉพาะแก๊ส He
ไหลผ่านคอลัมน์ที่บรรจุ
TiO2
เอาไว้
แล้วทำการเพิ่มอุณหภูมิและลดอุณหภูมิสลับกัน
๓ ครั้ง จะเห็นว่า TCD
ส่งสัญญาณออกมาเหมือนกับมีพีค
แต่ในความเป็นจริงนั้นสิ่งที่เห็นมีรูปร่างเหมือนพีคนั้นคือ
Base
line รายละเอียดของการทดลองนี้อ่านได้ใน
Memoir
ปีที่
๙ ฉบับที่ ๑๒๕๖ วันศุกร์ที่
๑๔ ตุลาคม ๒๕๕๙ เรื่อง "NH3-TPD การลาก base line (๒)"
สาเหตุที่
TCD
ส่งสัญญาณออกมาก็เพราะเมื่ออุณหภูมิคอลัมน์สูงขึ้น
แก๊สจะมีความหนืดมากขึ้น
จึงไหลผ่านเบดตัวอย่างที่บรรจุอยู่ในคอลัมน์ได้ยากขึ้น
ในกรณีนี้ถ้าเราวัดความดันด้านขาเข้าเบดเราจะเห็นว่าความดันด้านขาเข้าสูงขึ้น
อัตราการไหลโดยปริมาตรของแก๊ส
(ค่าที่ความดันด้านขาเข้าเบด)
จะลดต่ำลง
ความเร็วแก๊สที่ไหลผ่านขดลวดอ้างอิงก็ลดต่ำลงไปด้วย
ในขณะเดียวกันแก๊สอุณหภูมิสูงที่ไหลผ่านเบดตัวอย่างออกมานั้น
เมื่อไหลมาถึงตัวขดลวดฝั่งด้านที่เป็นตัววัด
ก็อาจมีอุณหภูมิสูงกว่าเดิม
ปัจจัยเหล่านี้ส่งผลให้
TCD
ส่งสัญญาณว่ามีการเปลี่ยนแปลงเกิดขึ้น
แต่การเปลี่ยนแปลงนี้ไม่ได้เกิดจากการเปลี่ยนแปลงองค์ประกอบของแก๊สที่ไหลเข้าตัว
TCD
แต่เกิดจาก
"ความเร็วของการไหล"
และ
"อุณหภูมิ"
ของแก๊สที่ไหลเข้าตัว
TCD
นั้นเปลี่ยนไป
NH3-TPD
เป็นเทคนิคที่นิยมใช้กันในการวัดปริมาณและความแรงของตำแหน่งที่เป็นกรดบนพื้นผิวของแข็งที่เป็นตัวเร่งปฏิกิริยา
ในการวิเคราะห์นี้จะมีการเตรียมตัวอย่างด้วยการให้ความร้อนแก่ตัวอย่าง
ณ อุณหภูมิหนึ่งนานเป็นช่วงเวลาหนึ่งก่อนภายใต้บรรยากาศแก๊สเฉื่อยเช่น
He
ขั้นตอนนี้ทำเพื่อกำจัดแก๊สอื่นที่ไม่ใช่
He
ออกจากรูพรุนของตัวอย่าง
จากนั้นจึงค่อยลดอุณหภูมิตัวอย่างแล้วให้ตัวอย่างดูดซับแก๊ส
NH3
จนอิ่มตัว
ตามด้วยการไล่แก๊ส NH3
ที่ไม่ถูกดูดซับด้วยการ
purge
ด้วย
He
ซ้ำ
ซึ่งเมื่อไล่ NH3
ที่ไม่ถูกดูดซับออกไปจนหมดแล้ว
แก๊สที่ไหลเข้า TCD
ฝั่งอ้างอิงและแก๊สที่ไหลผ่านตัวอย่างมาเข้า
TCD
ก็จะมีแต่
He
เป็นองค์ประกอบเท่านั้น

รูปที่
๒ การเปลี่ยนแปลงอุณหภูมิและสัญญาณที่
TCD
ส่งออกมา
เมื่อให้เฉพาะแก๊ส He
ไหลผ่านตัวอย่าง
TiO2
และทำการเพิ่มอุณหภูมิและลดอุณหภูมิสลับกับ
๓ ครั้ง
เส้นสีแดงที่เห็นว่ามีรูปร่างเหมือนพีคนั้นแท้จริงคือ
Base
line
แต่ในความเป็นจริงนั้นมีหลายปัจจัยด้วยกันที่ส่งผลให้สัญญาณที่
TCD
ส่งออกมานั้นไม่ได้เกิดขึ้นจากการมี
NH3
หลุดออกมาจากพื้นผิวเท่านั้น
แต่เกิดจากปัจจัยอื่นร่วมด้วย
เช่น ความเร็วและอุณหภูมิของแก๊ส
He
ที่ไหลเข้าตัว
TCD
ที่เปลี่ยนแปลงไปด้วยสาเหตุที่ได้กล่าวมาข้างต้น
และการใช้อุณหภูมิที่ไม่สูงพอและระยะเวลาที่ไม่นานพอที่จะไล่แก๊สอื่นที่ไม่ใช่
He
ออกจากรูพรุนของตัวอย่างจนหมดในขั้นตอนการเตรียมตัวอย่าง
ทำให้แก๊สเหล่านี้หลุดออกจากรูพรุนของตัวอย่างเมื่อเพิ่มอุณหภูมิตัวอย่างจนสูงมากพอ
การไล่แก๊สตรงนี้เป็นการไล่
"แก๊สชนิดอื่น"
ไม่ใช่เฉพาะ
"ความชื้น"
แบบที่ใครต่อใครชอบคิดกัน
ด้วยเหตุนี้การคำนวณปริมาณแก๊ส
NH3
ที่ตัวอย่างคายออกมานั้นโดยอาศัยสัญญาณ
TCD
ที่วัดได้จึงควรต้องใช้ความระมัดระวังมาก
เรื่องเหล่านี้เคยเล่าไว้บ้างแล้วใน
Memoir
ฉบับก่อนหน้าดังนี้
ปีที่
๓ ฉบับที่ ๒๖๔ วันอาทิตย์ที่
๒๗ กุมภาพันธ์ ๒๕๕๔ เรื่อง
"NH3-TPD - การไล่น้ำและการวาดกราฟข้อมูล"
ปีที่
๓ ฉบับที่ ๒๖๗ วันจันทร์ที่
๗ มีนาคม ๒๕๕๔ เรื่อง "NH3-TPD - การลาก base line"
ปีที่
๖ ฉบับที่ ๗๔๓ วันพุธที่ ๕
กุมภาพันธ์ ๒๕๕๗ เรื่อง
"NH3-TPD ตอน ตัวอย่างผลการวิเคราะห์ ๑"
กราฟผลการวิเคราะห์
NH3-TPD
ที่มีรายงานทั่วไปในบทความวิชาการต่าง
ๆ นั้น
ไม่ได้มีการระบุเอาไว้ว่าเป็นกราฟข้อมูลดิบที่ได้จริงจากการวัดหรือเป็นกราฟที่ผ่านการตัด
base
line ทิ้งแล้ว
โดยส่วนตัวแล้วเห็นว่าผล
NH3-TPD
จำนวนไม่น้อยที่มีการรายงานกันนั้นเป็นผลที่เกิดจากการตัด
base
line ทิ้งไปแล้วเพื่อทำให้เส้นกราฟดูดีโดยทำให้เหมือนกับว่าสัญญาณ
base
line นั้นขนานไปกับแกน
x
แต่ก็มีปรากฏในหลายบทความเช่นกันที่นำเสนอกราฟที่ยังไม่ผ่านการตัด
base
line
ซึ่งถ้าเราพิจารณาตัวเส้นกราฟกับข้อมูลตัวเลขที่เขารายงานเอาไว้นั้น
ก็พอจะมองเห็นความขัดแย้งอยู่
เริ่มจากตัวอย่างในรูปที่
๓ และ ๔ กราฟในรูปที่ ๓
นั้นมีการขยับเส้นไม่ให้ซ้อนทับกัน
ส่วนกราฟในรูปที่ ๔
นั้นเริ่มต้นโดยนำตำแหน่งสัญญาณเริ่มต้นการวัดมาไว้ที่ระดับเดียวกัน
จากกราฟทั้งสองเห็นได้ชัดว่าตำแหน่งเริ่มต้นของสัญญาณเมื่อเริ่มการวัดกับตำแหน่งสัญญาณเมื่อสิ้นสุดการวัดนั้นอยูคนละตำแหน่งกัน
โดยตำแหน่งเมื่อสิ้นสุดการวัดนั้นอยู่
"สูงกว่า"
ตำแหน่งเมื่อเริ่มต้นการวัด
การที่เห็นตำแหน่งสิ้นสุดการวัดนั้นอยู่
"สูงกว่า"
ตำแหน่งเมื่อเริ่มต้นการวัดมันก็แปลได้สองทาง
คือ (ก)
ยังมีการคายซับ
NH3
ออกมาอยู่
แต่หยุดการวิเคราะห์ก่อนที่จะคายออกมาหมด
ถ้าเป็นเช่นนี้ปริมาณตำแหน่งกรดที่วัดได้ก็ไม่ใช่ปริมาณ
"ทั้งหมด"
หรือ
(ข)
base line มีการเปลี่ยนตำแหน่ง
ถ้าเป็นเช่นนี้คำถามที่ตามมาก็คือ
base
line มีการเปลี่ยนแปลงรูปแบบใด
ซึ่งการเปลี่ยนแปลงนั้นไม่จำเป็นต้องเป็นเส้นตรงเสมอไป

รูปที่
๓ กราฟ NH3-TPD
ที่ใช้
TCD
เป็นตัวตรวจวัด
ในรูปนี้มีการขยับเส้นกราฟแต่ละเส้นไม่ให้ซ้อนทับกัน
ถ้าเป็นคุณ คุณจะลากเส้น
base
line โดยใช้แนวเส้น
1
หรือ
2
หรือจะลากเป็นอย่างอื่น

รูปที่
๔ อีกตัวอย่างของกราฟ
NH3-TPD
ที่ใช้
TCD
เป็นตัวตรวจวัด
ในบทความนี้เขียนกราฟโดยให้มีจุดเริ่มต้นอยู่ที่ระดับเดียวกัน
พึงสังเกตว่าแม้ว่าจะทำการวัดด้วยสภาวะเดียวกัน
แต่ตำแหน่งจุดสิ้นสุดของกราฟนั้นอยู่ที่ระดับที่ต่างกัน
ในกรณีเช่นนี้ถ้าเป็นคุณ
คุณจะลาก base
line เพื่อคำนวณปริมาณ
NH3
ที่ตัวอย่างคายออกมาอย่างไร
อีกประเด็นที่น่าสนใจก็คือ
ผลการวิเคราะห์ในรูปที่ ๓
และ ๔ ได้มาจากตัวอย่างที่มี
Al2O3
เป็นองค์ประกอบหลัก
และตัว Al2O3
ก็มีความเป็นกรดอยู่บนพื้นผิวด้วย
(บทความของคณะวิจัยในกลุ่มทำงานเดียวกัน)
แต่เมื่อเริ่มทำการวิเคราะห์ที่อุณหภูมิเริ่มต้นต่างกัน
(รูปที่
๓ เริ่มที่ 100ºC
ในขณะที่รูปที่
๔ เริ่มที่ 30ºC)
ลักษณะการปรากฏของพีคแรกที่อุณหภูมิต่ำนั้นแตกต่างกัน
โดยในรูปที่ ๓
นั้นจะเห็นว่าการเพิ่มขึ้นของสัญญาณจะเกิดขึ้นหลังจากที่อุณหภูมิสูงเกิน
100ºC
ได้ระดับหนึ่ง
(เห็นได้จากการที่เส้นกราฟค่อนข้างราบในช่วงแรกก่อนไต่ขึ้น)
นั่นแสดงว่าถ้าการเพิ่มขึ้นนั้นเกิดจาก
NH3
ที่พื้นผิวปลดปล่อยออกมา
การปลดปล่อยนั้นจะเริ่มเมื่ออุณหภูมิสูงเกิน
100ºC
ได้ระดับหนึ่ง
แต่ในรูปที่ ๔
นั้นจะเห็นว่าสัญญาณมีการเพิ่มสูงขึ้นตั้งแต่เมื่อเริ่มเพิ่มอุณหภูมิ
และมีสัญญาณออกมาเรื่อย ๆ
นั่นแสดงว่าถ้าการเพิ่มขึ้นนั้นเกิดจาก
NH3
ที่พื้นผิวปลดปล่อยออกมา
การปลดปล่อยนั้นจะมีอยู่ตลอดเวลาเมื่ออุณหภูมิของตัวอย่างสูงเกินกว่า
30ºC
ความแตกต่างของการทดลองทั้งสองอยู่ตรงที่การไล่
NH3
ที่ไม่ถูกดูดซับออกจากรูพรุน
โดยในรูปที่ ๓ นั้นทำการดูดซับที่อุณหภูมิ
100ºC
และทำการไล่ที่อุณหภูมิดังกล่าวนาน
1
ชั่วโมง
แต่ในรูปที่ ๔ นั้นทำการดูดซับที่อุณหภูมิ
30ºC
และทำการไล่ที่อุณหภูมิดังกล่าวนาน
3
ชั่วโมง
สาเหตุหนึ่งที่อาจเป็นไปได้ก็คือ
(ถ้าไม่คำนึงเรื่อง
base
line เปลี่ยนเนื่องจากการเปลี่ยนแปลงอุณหภูมิ)
การดูดซับที่อุณหภูมิสูงนั้นมี
NH3
ตกค้างน้อยกว่า
(แก๊สร้อนมีความหนาแน่นต่ำกว่าแก๊สเย็น)
และมีอัตราการแพร่ที่สูงกว่า
ทำให้การใช้เวลาเพียง 1
ชั่วโมงก็สามารถไล่
NH3
ที่ตกค้างอยู่ในรูพรุนออกได้หมด
เมื่อเริ่มเพิ่มอุณหภูมิตัวอย่างจึงไม่เห็นการเปลี่ยนแปลง
แต่การดูดซับที่ 30ºC
แม้ว่าจะทำการไล่
NH3
ไม่ถูกดูดซับออกจากรูพรุนนานถึง
3
ชั่วโมงก็อาจจะยังไล่
NH3
ที่ค้างอยู่ในรูพรุนออกได้หมด
ดังนั้นเมื่อทำการเพิ่มอุณหภูมิ
แก๊สที่ร้อนขึ้นมีการขยายตัว
โมเลกุล NH3
ที่ไม่ถูกดูดซับแต่ยังคงค้างอยู่ในรูพรุนก็เลยแพร่ออกมา
กลายเป็นสัญญาณให้เห็น
แต่สัญญาณนี้ไม่ควรตีความว่าเป็นการปลดปล่อย
NH3
ออกจากตำแหน่งที่เป็นกรด
แต่เป็น NH3
ในเฟสแก๊สที่ค้างอยู่ในรูพรุน
ในกรณีของตัวอย่างในรูปที่
๕ นั้น ขอให้ลองสังเกตเส้น
a
และ
e
ตรงที่ทำเครื่องหมายเลข
1
เอาไว้
บทความนี้อ่านสัญญาณ TCD
ที่เพิ่มขึ้นเมื่ออุณหภูมิเพิ่มสูงจาก
100ºC
ว่าเป็นสัญญาณที่เกิดจากการคายซับ
NH3
จากตำแหน่งที่เป็นกรดที่มีความแรงต่ำ
แต่กลับไม่อ่านสัญญาณ TCD
ที่เพิ่มขึ้นเมื่ออุณหภูมิอยู่ในช่วงระหว่าง
450-500ºC
ว่าเป็นสัญญาณที่เกิดจากการคายซับ
NH3
จากตำแหน่งที่เป็นกรดที่มีความแรงสูง
ในส่วนของวิธีการทดลองนั้นบทความก็ระบุเอาไว้ชัดเจนว่าในขั้นตอนการให้ความร้อนไล่
NH3
ออกจากพื้นผิวนั้นใช้อุณหภูมิสูงถึง
700ºC
แต่กราฟที่นำมาแสดงกลับตัดมาแค่อุณหภูมิประมาณ
550ºC
เท่านั้นเอง
จึงทำให้มองไม่เห็นว่าที่อุณหภูมิสูงกว่า
550ºC
นั้น
สัญญาณของกราฟทุกเส้น
โดยเฉพาะเส้น a
e f และ
h
นั้นมีการไต่ขึ้นไปสูงแค่ไหน
หรือในกรณีของเส้น
e
f g และ
h
ตรงตำแหน่งที่ทำเครื่องหมายเลข
2
เอาไว้
ในบทความนั้นอ่านว่าเส้น
f
และ
g
มีพีคที่เกิดจากการคายซับ
NH3
จากตำแหน่งที่เป็นกรดที่มีความแรงสูง
แต่ในกรณีของเส้น e
และ
h
นั้น
จะว่าไปแล้วเส้น e
มีลักษณะที่เป็นพีคที่เด่นชัดกว่าของเส้น
h
แต่บทความกลับอ่านว่าเส้น
e
ไม่มีพีคของตำแหน่งที่เป็นกรดที่มีความแรงสูง
แต่ของเส้น h
กลับอ่านว่ามี
โดยส่วนตัวแล้วจะใช้การวัดปริมาณเบสที่ตัวอย่าง
"ดูดซับ
(adsorption)"
เอาไว้ได้ควบคู่ไปกับผล
NH3-TPD
เพื่อใช้ประกอบการพิจารณาว่าตำแหน่งไหนที่เป็นพีคที่แท้จริง
และปริมาณ NH3
ที่ไล่ออกมานั้นออกมาหมดแล้วหรือยัง
เพราะการดูผลการ "คายซับ
(desorption)"
เพียงอย่างเดียวไม่สามารถบอกได้ว่าได้ใช้อุณหภูมิที่สูงพอที่สามารถไล่
NH3
ออกจากพื้นผิวได้หมดหรือไม่
อันที่จริงยังมีเรื่องของ
H2-TPR
เตรียมไว้อีก
แต่ฉบับนี้รู้สึกว่าจะยาวพอสมควรแล้ว
เลยต้องขอพักไว้ตรงนี้ก่อน


รูปที่
๕ กราฟ NH3-TPD
และปริมาณตำแหน่งที่เป็นกรดที่ทางคณะผู้วิจัยคำนวณไว้
การวัดนี้ใช้ TCD
เป็นตัวตรวจวัด
พึงสังเกตบริเวณตัวอย่างที่ใส่หมายเลข
(1)
และ
(2)
เอาไว้